대부분
파브리병은 GLA 유전자의 돌연변이로 인해 발생하는 희귀 유전 질환입니다.

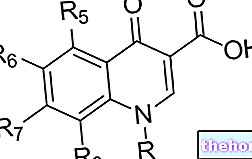
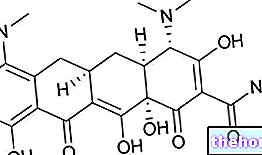
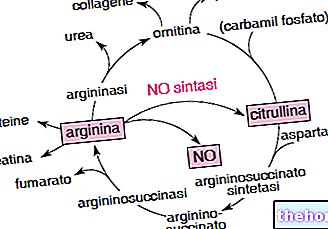
수치: 알파갈락토시다아제 A의 구조
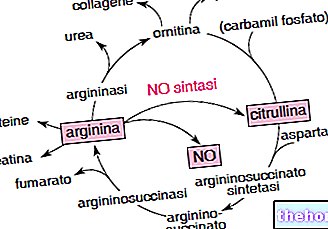
GLA 유전자는 X 염색체에 위치하며 알파-갈락토시다제 A라는 효소를 암호화합니다. 이 효소는 글로보트리에소실세라마이드로 알려진 지질 분해에 중요한 역할을 합니다.
파브리병 환자의 경우 알파-갈락토시다아제 A 효소 기능이 제대로 작동하지 않아 글로보트리에소실세라마이드 분자가 특정 세포 내 소기관(리소좀)에 비정상적으로 축적되는 경향이 있어 영향을 받은 세포에 심각한 고통을 줍니다.
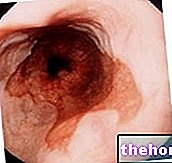
파브리병은 신경계, 피부과, 안과, 위장, 뇌혈관, 신장 및 심장 임상 증상을 유발합니다.
절대적으로 파브리 증후군을 진단하기 위해서는 적절한 유전자 검사가 필수적입니다.
현재로서는 파브리 증후군을 구체적으로 치료할 수 있는 치료법이 없고 증상이 있는 치료(즉, 증상 완화를 목표로 함)만 있습니다.
이러한 증상 치료 중 가장 중요한 것은 실험실에서 만든 알파-갈락토시다제 A 효소 유사체를 투여하는 효소 대체 요법입니다.
파브리병이란?
파브리병은 글로보트리에소실세라마이드(globotriesosylceramide)라고 하는 특정 유형의 지질이 혈관, 조직 및 기관의 벽에 축적되어 발생하는 유전성 유전 질환입니다.
파브리병은 스핑고지질증이며 소위 리소좀 축적 질환의 이질적인 그룹에 속합니다.